 协同生物
协同生物译者导读:这是一篇关于构建小鼠胆结石疾病模型以研究人类胆结石形成机制的文章,该文论述了小鼠胆结石模型建立的研究历程,指出了人类胆结石形成机制的新思路。
重点是,文章总结了诸多研究的过程与结果,这些研究通过结合基因组学方法和小鼠表型研究成功鉴定了Lith基因,为人类Lith基因的发现推平道路。用基因敲除或转基因方法为胆结石这种“古老而又新鲜”的肝胆疾病的病理生理学研究提供了新的视角。对于从事胆结石课题研究、动物模型建立、高胆固醇饲料制备等工作的研究者来说,此文值得一读。
作者:Tony Y. Wang、Piero Portincasa等
译者:协同生物苏永腾博士
前言
胆结石是最常见的肝胆疾病之一,12%美国成年人深受其害,为减少胆结石的发病率、死亡率,深入研究胆结石的病理生理学将有助于开发出创新有效的治疗方法。
从20世纪50年代开始,研究者就通过给仓鼠、老鼠、豚鼠、兔子和猴子等实验动物喂养数周到几个月的高胆固醇饲料并成功建立了胆结石模型,从而根据模型研究胆结石疾病的发病机制。直至上世纪90年代,第一个与胆结石相关的基因-Lith1在近交系C57L/J小鼠中得以鉴定。
我们通过此文介绍小鼠胆结石模型的建立、模型对遗传研究以及胆结石病理生理学研究的贡献,为胆结石研究做一个概览综述。
小鼠胆结石模型的建立
Tepperman等研究者率先发现可以通过胆固醇和胆酸的组合使用来建立小鼠胆结石模型。在喂养含有胆固醇和胆酸的饲料后,发现从小鼠胆石中提取出来的甾醇基本上是胆固醇,占了结石总重量的94%,而单独喂养1%胆固醇饲料2-8个月却不能诱发出胆结石。由此可见,胆酸在小鼠胆结石模型建立中必不可少。
1978年,Fujihira等研究者给七种近交系小鼠饲喂高胆固醇饲料8周后观察到小鼠出现胆结石,患病率从0%到100%不等。Alexander和Portman等人的研究进一步确认了小鼠在高胆固醇饲料的喂养下,胆结石患病率存在品系差异,这些研究分析为小鼠胆结石病基因研究打下了良好基础。
更重要的是,Wang等人发现小鼠喂养高胆固醇饲料8周内胆固醇结晶和胆结石形成的具体过程,先是粘蛋白凝胶的初始积累,继而出现无水或液晶状外观,然后这些胆固醇晶体聚集,最后出现砂质结石和真胆结石(见下图)。

胆结石易感小鼠C57L/J饲喂高胆固醇饲料8周内胆固醇结晶和胆结石形成的显微照片(用相衬法,偏光显微镜)
(a)粘蛋白凝胶;(b)小液晶;(c)聚集液晶;(d)具有麦芽交叉双折射和焦点圆锥结构的熔融液晶;(e)弧形(可能是无水胆固醇)晶体;(f)丝状晶体;(g)端部管状晶体破裂产生胆固醇一水合物晶体;(h)典型的胆固醇一水合物晶体,从28和100.88的角度;(i)聚集的胆固醇一水合物晶体;(j和k)砂质结石;(l)胆结石。
小鼠模型对胆结石遗传学研究的贡献
流行病学和临床调查(如对不同家庭和双胞胎的相关调查)研究已很清楚地表明了遗传基础在个体患胆结石病方面发挥了关键性的作用。然而,人们还没有全部掌握遗传因素对胆结石的影响,因为胆结石是由多种环境因素以及未知基因相互作用和复杂调控引起的。
此外,为了定量研究多基因性状,常规单基因性状遗传作图法不适用于胆结石研究。近交系老鼠有着优良的遗传资源,是研究胆结石遗传学的优选动物模型。Khanuja等人在给实验小鼠饲喂高胆固醇饲料12周后,率先发现了胆结石易感鼠C57L/J和抗胆结石鼠AKR/J之间成石的差异是由两个Lith基因决定——Lith1和Lith2.
随后,C57L/J和AKR/J小鼠的肝、胆囊、小肠等部位都被用来进行了系统的基因表型对比研究。为了判断Lith1和Lith2两个基因中哪个单独诱导胆结石的产生,携带Lith1的C57L/J小鼠(胆结石易感)或Lith2等位基因的AKR/J小鼠(抗胆结石)模型被建立并进行致石研究,结果发现Lith1和Lith2通过不同的成石机制诱导胆结石的发生,Lith1调控胆固醇的代谢,Lith2则与胆汁酸分泌密切相关。
研究者通过更多的近交系小鼠胆结石模型研究发现,还有更多Lith基因参与胆结石的成石调控,Lith基因家族Lith1~Lith25在染色体上的分布如下图所示。

胆结石(Lith)基因图谱
一条垂直线代表一条染色体,着丝粒在顶端。距着丝粒(水平黑线)的遗传距离显示在染色体的左侧。胆结石QTL(Lith基因)和候选基因的位置用水平黑线表示,基因符号在右边(基因符号和名称略)。
因为人类和小鼠染色体同源的原因,以上这些来自小鼠的基因研究对人类的Lith基因的发现有着指导性的作用,并成功证实了人类的LITH基因ABCG5/G8。
研究者先是在小鼠身上发现和鉴定了ABCG5/G8基因,随后在德国人和智利人身上发现ABCG5/G8的两个相关变体(即ABCG5-R50C和ABCG8-D19H),之后又在中国人和印度人身上得以印证。
因此,根据小鼠的Lith基因图谱,人类的更多Lith基因及其致病性机制将得以发现和阐明。2010年,研究者首次提出了胆结石病的五种发病因素(见下图):遗传因素和Lith基因;肝胆固醇高分泌,导致胆固醇过饱(即高胆固醇饱和指数);胆固醇结晶和固体胆固醇晶体的生长;胆囊运动受损;肠道因素,包括吸收的胆固醇从小肠输送到肝脏引起胆汁高分泌,以及肠道微生物群的变化。
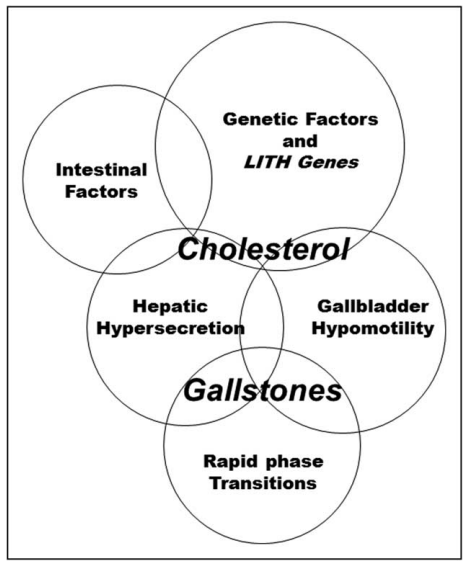
五种因素的相互作用示意图:(i)遗传因素和Lith基因;(ii)肝脏高分泌;(iii)胆囊动力低下;(iv)快速相变以及(v)肠道因素。持续性肝胆固醇高分泌是遗传因素调控的结果,导致胆固醇过饱和胆汁的形成,加速胆固醇结晶。胆囊运动功能受损可导致多余粘蛋白凝胶的产生和积累,促进胆泥的形成和微小结石的生长。这些改变也破坏了胆汁酸(肠道因素)的肝肠循环动力学,导致肠道吸收和胆汁酸池的变化。胆固醇吸收的增加将过量胆固醇输送到肝脏,分泌到胆汁中。肠道微生物群的异常可能会破坏肝脏、肠道和胆汁中胆固醇和胆汁酸的代谢,并损害胆囊的排空和再灌注。
可见,胆固醇晶体的快速生长、聚集形成微小结石、最终成石这一结果都是由于胆固醇过饱和导致的,还有就是胆囊粘液的高分泌以及胆囊受损的凝胶形成,最终导致了胆泥(即胆结石的前体物)的形成。
胆结石病理生理学研究最新进展
研究发现女性胆结石患病率明显较高于同年龄段的男性,这意味着雌激素在胆结石的发病机制中发挥了关键性的影响。尽管雌激素受体a(ERa)能通过调控雌二醇(E2)的形成来诱导雌性小鼠胆结石的发生,但E2对小鼠的致石机理尚不明确。
一些研究发现,ERa基因敲除小鼠大剂量E2处理后仍发现胆结石。此外,有人发现名为Lith18G的一种胆结石基因同时也是一种雌激素受体——G蛋白偶联受体30(GPR30)。这一发现带来了一个新问题,E2是如何通过有效绑定并激活GPR30和ERa来影响胆结石的形成呢?
为了区分ERa和GPR30的致石作用,需要对整个胆固醇的结晶路径和顺序去探究。通过雌性野生型卵巢切除、GPR30敲除、ERa敲除和GPR30/ERa双基因敲除等方法进行分析研究。E2分别与GPR30以及ERa结合后,激活了液晶和无水晶体的产生,使得胆汁从过饱和状态进化到中间体结晶物。
此外,在GPR30/ERa-DKO小鼠模型中,胆固醇的结晶明显受阻。这表明GPR30与ERa在促进E2诱导的胆结石发生中具有协同作用。由于GPR30主要存在于肝细胞的内质网而不是细胞核内,其可能是通过表皮生长因子受体信号级联途径抑制肝脏胆固醇7a羟化酶和胆汁酸的合成,导致肝脏过量分泌胆固醇并成石。
非酒精性脂肪肝(NAFLD)是胆结石形成的一个重要影响因素,NAFLD与胆结石之间的联系机制仍不清楚。缺氧诱导因子1(HIF1)是调节氧传递、细胞生长和氧化还原稳态相关基因表达的重要转录因子,促进机体对缺氧环境的适应性反应。
HIF1A主要存在于肝脏的周围静脉,即生理性缺氧的区域中。肝脏脂肪变性时,脂质的积累使肝细胞的体积明显增大,循环减少而最终导致缺氧。Asai等研究发现,在HIF1A基因敲除小鼠中,一种负责将肝脏分泌液分泌到胆管的水通道蛋白-8(AQP-8)的表达显著增加。这些变化使胆汁流量显著增加35%,胆囊和肝脏胆汁中的胆汁脂质浓度降低36%,并减轻了胆囊的炎症。
结果,肝脏特异性HIF1A基因敲除小鼠的胆固醇结晶被抑制,胆结石的形成也被阻止。相反,在饮食诱导的肝脂肪变性中,由于HIF1A通路的激活,野生型小鼠肝脏加速了胆结石的形成。此外,在伴有胆结石的NAFLD患者肝脏中HIF1A及其下游靶点的表达增加,这种结果提示了HIF1A在NAFLD患者的胆结石中发挥了重要影响。胆汁的流动是由胆汁酸依赖性和非胆汁酸依赖性两种途径决定,水和离子/非离子溶质通过跨细胞和细胞旁途径经肝细胞的转运促使了胆汁的形成。
Cludin 2(Cldn2)是一种细胞旁通道形成蛋白,在肝细胞和胆管细胞中高度表达。Matsumoto研究发现Cldn2基因敲除小鼠的胆汁流速与野生型小鼠相比降低了50%,这是由于肝胆系统的水渗透性降低所致,从而显著增加胆汁中的总脂质浓度。4周后,Cldn2基因敲除小鼠产生了胆结石,而野生型小鼠并没有出现结石,这表明Cldn2对胆汁形成和胆汁脂质稳态至关重要。
有研究发现,在ABCG5和ABCG8两种基因单独缺失或都缺失的小鼠肝脏中胆固醇分泌显著降低,但没有完全消除,这表明存在一种ABCG5/G8非依赖通路可调节人体和小鼠的胆汁胆固醇分泌。
Wang等研究者指出,在成石状态下,ABCG5/G8非依赖通路胆固醇输出占总量的30%至40%,在高胆固醇饮食中调节机体胆固醇分泌上起着关键作用。在ABCG5/G8缺失的情况下,这种途径参与了胆固醇的分泌和胆结石形成。与ABCG5/G8不同的是,这种通路途径并不是由LXR通过其信号通路激活的。这一发现表明了ABCG5/G8依赖性和ABCG5/G8非依赖性通路在肝脏胆固醇分泌和胆囊结石形成中都发挥着重要作用。
临床研究发现,肠道胆囊收缩素(CCK)的分泌和胆囊排空受高脂饮食的破坏,但腹腔疾病对胆结石形成的影响目前很少有研究。Wang等人建立CCK缺乏小鼠模型来研究腹腔疾病对胆汁分泌的影响,发现CCK基因敲除后小鼠表现胆囊排空受损,小肠转运时间减少,肠道胆固醇吸收增加,过量的肠道源性胆固醇导致胆汁高分泌,从而引起胆固醇结晶和胆石形成。此外,有研究发现强效CCK-1受体拮抗剂Devaxipide通过损害小鼠胆囊排空功能,扰乱胆固醇代谢和增强肠道胆固醇吸收,增加了胆结石形成的风险。上述实验为研究腹腔疾病患者胆结石的发病机制提供了重要依据。
胆汁中可能同时含有促核因子和抗核因子,其含量的不平衡可能导致胆固醇快速结晶从而形成结石,但这一说法仍需要更多的研究来证实。粘蛋白是第一个被发现的促进胆固醇结晶成石的蛋白分子,还有许多其他蛋白也被认为是影响胆汁中胆固醇结晶的促核因子或抗核因子,但它们作用机制仍不清楚。
Niemann Pick C2(NPC2)是一种可溶性溶酶体蛋白,通过激活ABCG5/G8介导的胆固醇转运通路来调节肝脏胆固醇分泌,它促进转基因小鼠(人NPC2基因)的胆固醇结晶和胆石形成。相比而言,NPC2基因缺乏的小鼠在高胆固醇饲料饲喂2周后肝脏胆固醇分泌和胆石率减少。上述结果表明,这些胆汁蛋白可能通过胆汁中的液晶途径来调节胆固醇的结晶以导致胆结石发生。
肠道胆固醇的高效率吸收和高胆固醇饮食(西方饮食模式)是小鼠的两个致石因素。深入研究发现,有很多因素都可以在肠道胆固醇吸收、运送致肝脏并分泌于胆汁中的途径上影响胆固醇的结晶而导致胆结石的形成,如胆固醇酯化、肠细胞内脂质转运、乳糜微粒的聚集等。此外,肠道微生物群和免疫系统对胆结石形成也有重要的影响,但仍待进一步深入研究。
因此,那些影响肝细胞转运和胆汁分泌胆固醇、磷脂和胆汁酸的因素都可以通过引起胆固醇高分泌和胆固醇过饱和胆汁来促进胆结石的形成。一些受体,如liver X receptor (LXR)、farnesoid X receptor(FXR)、pregnane X receptor (PXR)、constitutive androstane receptor (CAR)、 ERa等可以通过肝脏脂质代谢的途径来影响胆结石的形成。还有,对肝脏胰岛素受体的破坏也能提高小鼠胆结石的发病率,这种破坏可以降低胆固醇7a羟化酶的活性并影响胆汁酸的合成,但胰岛素抵抗在胆结石形成中的作用机制也尚不明确。
结论
上述小鼠胆结石研究的成果为研究同源人类结石基因和探索人类胆石形成的机制打开了大门。在未来的研究中,小鼠模型建立和胆结石研究和应聚焦于核受体、肠道微生物群、血脂、高胰岛素血症、非酒精性脂肪肝、肥胖、糖尿病、衰老以及不运动的生活方式等方面对胆结石形成的影响。这些小鼠模型实验将为肝胆疾病的治疗提供新的依据。
本文来源:Mouse Models Of Gallstone Disease;Tony Y. Wang/Piero Portincasa著






